Author: Nihal SARMAŞIK, Group Manager (Health & Transportation)
Today, medical device technology is growing fast for enhanced patient care and therapies. Wide variety of medical devices and systems have been developed over decades. These devices are indispensable part of diagnostics and treatment of diseases and they’re still evolving to enhance their functionality. Guided surgical tools, CAD’s, CT’s, MRI’s, X-Ray and fluoroscopy imaging systems, surgical robotics are examples of such devices. These consist of high technology and complex components. That sort of complexity requires more software-oriented solutions; and thus, software becomes a vital part of the systems.

Beside their complexity, these systems are safety-critical. That means any kind of failure or malfunction may result in serious injury or even death. For this particular reason, faults in these systems are main elements to be avoided for achieving an acceptable risk level. On the other hand, trends in healthcare show a path towards home-care and personalized usage of medical devices; meaning the usage extends to non-clinical environments. This alone increases the importance of safety and reliability. To cope with these challenges rigid set of rules must be applied, as shown in Figure-1. These rules are regulated by MDR 2017/745 (Medical Device Regulations) in Europe.
MDR lays down rules to ensure high standards of quality and safety for medical devices. This makes sure that a high level of protection is maintained for the health and safety of patients and users. MDR defines classification rules to identify a severity level for a medical device. Severity level depends on the harm on a patient in case of a device malfunction. MDR demands systematically defined and applied risk management process that works hand-in-hand with other disciplines. Being in the core of regulation and standards ecosystem, IEC 14971 precisely defines the application of risk management to medical devices. Risk management process plays an important role to identify safety class of the related software and its components. In addition to that, based on this safety class, the level of deliverables required for a certification is determined with the standard IEC 62304. This is a harmonized standard within EU (European Union) and it is also recognized by the FDA (Food and Drug Administration). IEC 62304 defines software life-cycle process for medical devices.
Usability engineering is another main concern for risk assessment. The standard of IEC 62366 defines a process to analyze, specify, develop and evaluate the usability of a medical device as it relates to safety. This process permits to assess and mitigate the risks associated with the “correct use” and “use errors”.
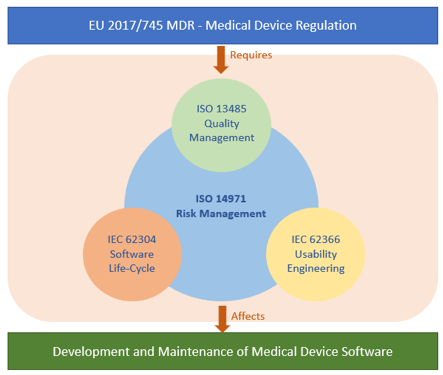
Figure-1: Relationship between medical device standards
All the standards above declare the requirements in a generic way to cover a wide range of medical devices and their software. For this reason, good understanding of these standards with a deep experience is essential to develop norm-compliant complex systems. In order to pass legitimate audits, one needs dedicated resources and time upon development.
Aside from all, time-to-market is a critical commercial issue. This puts great pressure on development teams. Even though the medical device development is highly regulated by industry standards, changes in the requirement set are quite often. Therefore, quick adaption to such changes is critical. Delivering solutions on time under high rate of requirement changes while ensuring safety is a great challenge. To overcome this challenge, new development approaches should be thought. The “V-Model”, widely used in avionics, automotive and healthcare domains, is an essential part of complex and safety-critical system development. To keep pace with this challenge, V-Model can also be leveraged by adapting agile paradigm while keeping safety in focus. A special hybrid process may be used where “V-model” is blended with the iterative and incremental methods, as shown in Figure-2. The main idea behind this mixture is to reevaluate design issues with respect to safety and reliability concerns at the end of each coding cycle.
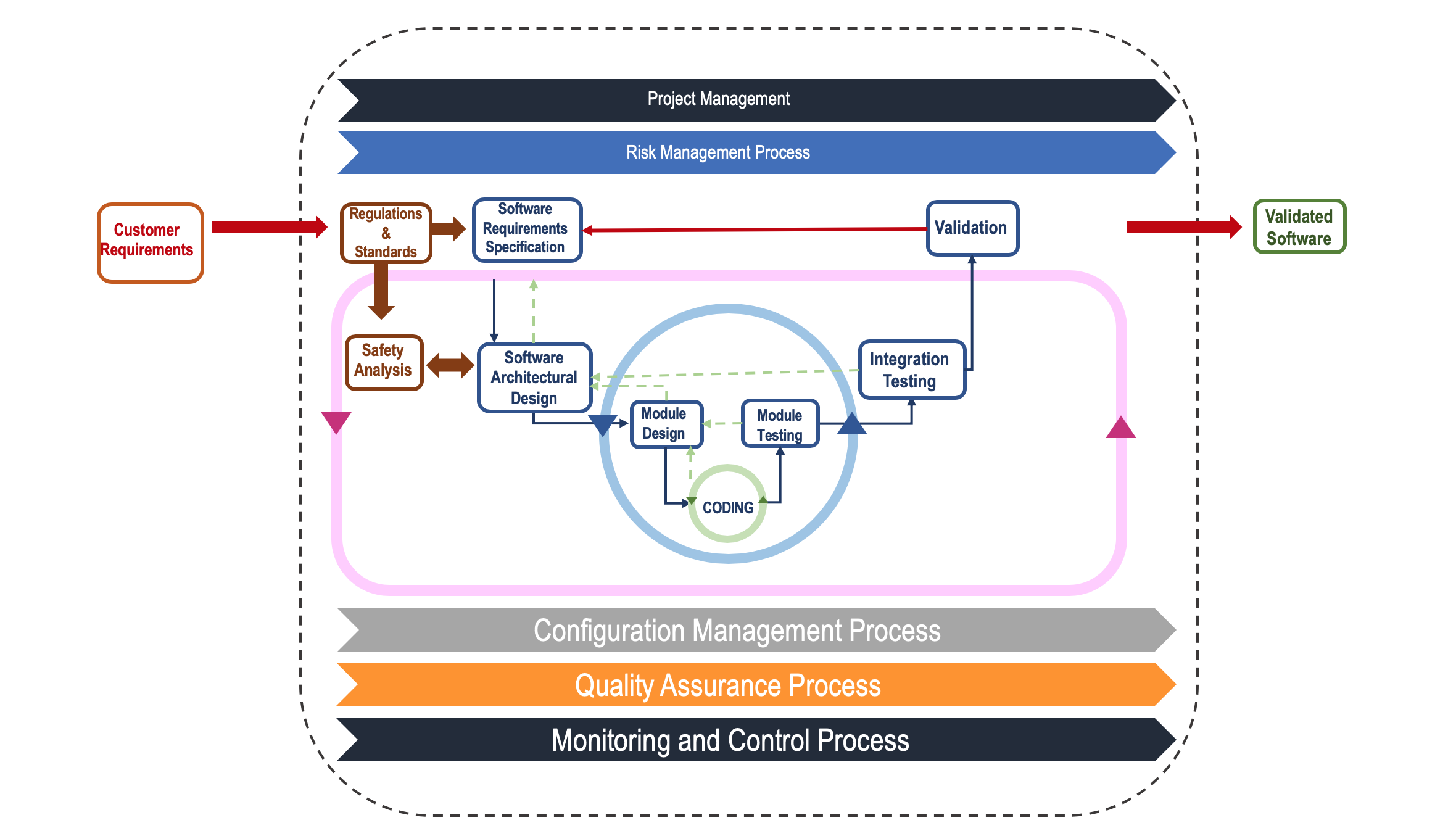
Figure-2: Hybrid V-Model
Marketing a medical device is subject to certification. As shown in Figure-3, certification requires that one provides acceptable proof to the authorities to show system requirements have been satisfied. To provide evidence systematically, all process artifacts and their dependencies should be linked. This can be done using bi-directional traceability. Traceability provides a systematic method to present acceptable proof to the authorities. There are various kinds of links between process artifacts and their dependencies. To handle this variety and to present this bi-directional traceability in a systematic manner, one requires a solid set of integrated tools chain.
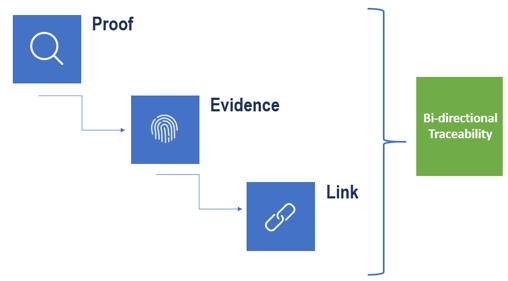
Figure-3: Bi-directional Traceability – Acceptable proof for certification
To summarize, software development for medical devices requires solid understanding of regulations and standards. Changing market dynamics, new challenges and technology should be considered at every stage of development. Before hitting this road, software development tools and techniques should be evaluated carefully, and processes must be established concisely.
References:
- EU 2017/745 Medical Device Regulations, the European Parliament and the Council of the European Union, 2017
- ISO 14971:2012 Medical Devices — Application of risk management to medical devices, The British Standards Institution, 2012
- BS EN 62304:2006 Medical Device Software — Software life-cycle processes, The British Standards Institution, 2008
- BS EN 62366-1:2015 Medical Devices Part 1: Application of usability engineering to medical devices, The British Standards Institution, 2015